Neglect of the internal degrees of freedom of the guest
The model calculations assume that the free energy contributions, due to the internal degrees of freedom of the guest, are not significantly affected in the act of adsorption.
In small, stiff molecules such as H2, only the ground vibrational level is expected to be populated near the room temperature.
As a result, changes in the internal degrees of freedom should be dominated by variations in the zero-point energy (ZPE) and molecular reorientation due to the anisotropy of the adsorption potential.
In order to illustrate the effect of these two terms, one can consider three examples of increasingly strong interaction between the guest (H2) and the host (C6H6 molecule; K+ and Mg^2+ ions).
In all three cases, H2 is coordinated at axially symmetric site.
The relevant energetic parameters[34] of the three complexes are summarised in Table 1.1.
Softening of the H2 vibrational mode upon adsorption changes the adsorption constant by
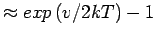 .
At 200K (kT=139cm
.
At 200K (kT=139cm ), the zero-point energy contribution would amount to 11% increase in the adsorption constant for H2 on benzene, and 17% increase for the ionic H2/K+ complex.
Both contributions are small compared to other sources of error in that treatment.
For most physisorption substrates based on covalently-bound solids the guest-host interactions are expected to fall in the same range.
For the much stronger bound H2/Mg^2+ complex, the zero-point energy (ZPE) contribution enhances the equilibrium
constant by a factor 5 at 200K, and a factor of 2.9 at the room temperature.
Therefore, the featureless particle approximation would not be appropriate for this system.
), the zero-point energy contribution would amount to 11% increase in the adsorption constant for H2 on benzene, and 17% increase for the ionic H2/K+ complex.
Both contributions are small compared to other sources of error in that treatment.
For most physisorption substrates based on covalently-bound solids the guest-host interactions are expected to fall in the same range.
For the much stronger bound H2/Mg^2+ complex, the zero-point energy (ZPE) contribution enhances the equilibrium
constant by a factor 5 at 200K, and a factor of 2.9 at the room temperature.
Therefore, the featureless particle approximation would not be appropriate for this system.
The hydrogen molecule possesses the largest known rotational constant 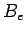 (60.853 cm),
so that the spacing of the rotational levels (
(60.853 cm),
so that the spacing of the rotational levels (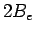 ) is comparable to
) is comparable to 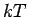 even at the room
temperature. Therefore, the estimation of the rotational contribution to the adsorption free
energy requires diagonalisation of the rotational Hamiltonian in the perturbing field.
In the rigid free rotor approximation, the rotational eigenfunctions of a homonuclear
diatomic molecule with nuclear spin
even at the room
temperature. Therefore, the estimation of the rotational contribution to the adsorption free
energy requires diagonalisation of the rotational Hamiltonian in the perturbing field.
In the rigid free rotor approximation, the rotational eigenfunctions of a homonuclear
diatomic molecule with nuclear spin 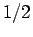 (such as ^1H2) are given by[42]:
(such as ^1H2) are given by[42]:
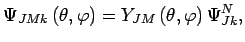 |
|
|
(1.12) |
where 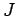 is the rotational angular momentum,
is the rotational angular momentum, 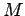 is its projection of the quantisation
axis,
is its projection of the quantisation
axis, 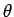 and
and 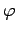 are polar angles,
are polar angles, 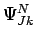 are nuclear spin eigenfunctions, and
are nuclear spin eigenfunctions, and 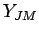 are spherical harmonics, given by:
are spherical harmonics, given by:
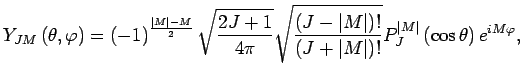 |
|
|
(1.13) |
where 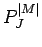 are the associated Legendre polynomials.
Quantum number can take all non-negative values, while is between
are the associated Legendre polynomials.
Quantum number can take all non-negative values, while is between 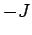 and . For even values of , a single symmetric nuclear spin wavefunction is allowed.
For odd , is antisymmetric and has three degenerate eigenfunctions.
The unperturbed eigenvalues corresponding to
and . For even values of , a single symmetric nuclear spin wavefunction is allowed.
For odd , is antisymmetric and has three degenerate eigenfunctions.
The unperturbed eigenvalues corresponding to  are given by:
are given by:
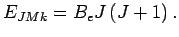 |
|
|
(1.14) |
An anisotropic real adsorbing potential
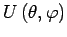 can be represented as an expansion over spherical harmonics:
can be represented as an expansion over spherical harmonics:
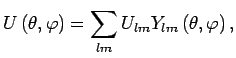 |
|
|
(1.15) |
where spherical components 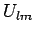 satisfy:
satisfy:
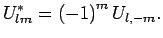 |
|
|
(1.16) |
For a homonuclear molecule, particle interchange symmetry requires that all contributions with 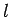 odd vanish.
Matrix elements of
can be evaluated explicitly in terms of the
odd vanish.
Matrix elements of
can be evaluated explicitly in terms of the 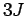 symbols (see ref. Landau1977):
symbols (see ref. Landau1977):
![$\displaystyle \jot9pt
\langle \Psi_{J'M'k'}\vert U\vert\Psi_{JMk}\rangle =\sum_...
...ac{\left(2l+l\right) \left(2J+l\right) \left(2J'+l\right) }{4\pi} \right]^{1/2}$](img115.png) |
|
|
|
![$\displaystyle \left[ \begin{array}{ccc}
J' & l & J \\
-M' & m & M\\
\end{arra...
...0 & 0\\
\end{array} \right]
\langle \Psi_{J'k'}^{N}\vert\Psi_{Jk}^{N}\rangle .$](img116.png) |
|
|
(1.17) |
These matrix elements vanish by symmetry unless 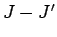 is even,
is even, 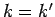 , and
, and 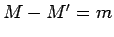 .
If the orientation of the local coordinate system is chosen to coincide with the principal
axes of the potential anisotropy tensor, the unique non-vanishing components of
for
.
If the orientation of the local coordinate system is chosen to coincide with the principal
axes of the potential anisotropy tensor, the unique non-vanishing components of
for 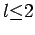 take a particularly simple form:
take a particularly simple form:
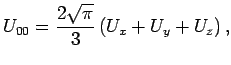 |
|
|
(1.18) |
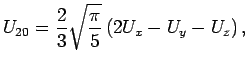 |
|
|
(1.19) |
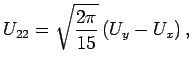 |
|
|
(1.20) |
where 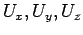 are the adsorption energies for the main axis of the guest oriented along the
are the adsorption energies for the main axis of the guest oriented along the 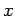 ,
, 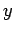 , and
, and 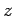 axes, respectively.
The
axes, respectively.
The 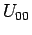 contribution does not mix the rotational eigenstates, and can be incorporated in the mass-particle solution.
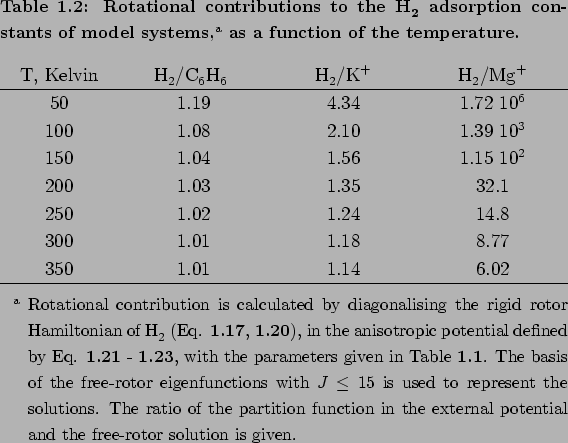
The effect of the anisotropic contributions of the adsorption constant is illustrated in Table 1.2[34].
contribution does not mix the rotational eigenstates, and can be incorporated in the mass-particle solution.
The effect of the anisotropic contributions of the adsorption constant is illustrated in Table 1.2[34].
For the low-anisotropy H2/C6H6 system (
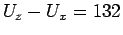 cm),
the neglect of rotational degrees of freedom does not become a significant source of
error until temperatures are below 100K. For the more anisotropic H2/K+ model
(
cm),
the neglect of rotational degrees of freedom does not become a significant source of
error until temperatures are below 100K. For the more anisotropic H2/K+ model
(
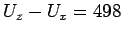 cm), featureless particle approximation provides useful estimates for the
adsorption constant at T
cm), featureless particle approximation provides useful estimates for the
adsorption constant at T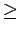 200K, but begins to break down at lower temperatures.
Finally, for the strongly anisotropic H2/Mg^2+ system (
200K, but begins to break down at lower temperatures.
Finally, for the strongly anisotropic H2/Mg^2+ system (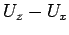 =2478cm,
or
=2478cm,
or 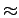 30 kJ
30 kJ mol),
rotational degrees of freedom cannot be neglected at any temperature of interest for
hydrogen storage applications.
mol),
rotational degrees of freedom cannot be neglected at any temperature of interest for
hydrogen storage applications.
Lyuben Zhechkov
2007-09-04