Many H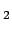 -PAH interactions
-PAH interactions
To assess the mutual influence and the highest possible density in a hydrogen monolayer adsorbed on graphene, models of PAHs with more than one H2 molecule have been calculated.
Two H2 molecules at opposite sides of a ring have about the same physisorption energy per H2, but slightly lower than for only one H2 molecule.
The only exception is  again triphenylene, which shows 0.3 kJ
again triphenylene, which shows 0.3 kJ mol
mol stronger interaction per H2 in the case where the central ring is covered on both sides.
For non-symmetric physisorption on top and bottom of the PAH, without exceptions the interaction energy of two H2 molecules, one above and one below a PAH (despite the site of adsorption), is found to be nearly the sum of the interaction energies of the single H2-PAH interaction.
However, the adjacent rings cannot be occupied with H2 at each ring at the same side, as the interaction between two H2 molecules is strongly repulsive at such distances (
stronger interaction per H2 in the case where the central ring is covered on both sides.
For non-symmetric physisorption on top and bottom of the PAH, without exceptions the interaction energy of two H2 molecules, one above and one below a PAH (despite the site of adsorption), is found to be nearly the sum of the interaction energies of the single H2-PAH interaction.
However, the adjacent rings cannot be occupied with H2 at each ring at the same side, as the interaction between two H2 molecules is strongly repulsive at such distances (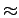 1.4 Å).
On the other hand, two H2 molecules show no repulsive interaction at a distance of the 2
1.4 Å).
On the other hand, two H2 molecules show no repulsive interaction at a distance of the 2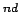 neighbour ring.
These observations are in agreement with Diep's and Johnson's study of the H2-H2 interaction [54,55].
Therefore, all geometrical models with several H2 molecules at a PAH side have the neighbouring rings of an H2 physisorption site uncovered.
Considering the above mentioned relations, one H2 molecule can be attached to each side of benzene and naphthalene (one at each side), two to each anthracene side, and three to each side of pentacene, triphenylene and coronene.
Table 2.2 shows that the total physisorption energy per H2 is the average of the individual physisorption energies of the occupied sites.
In other words, attaching H2 molecules at a PAH side is a completely additive process, and for a graphene sheet a constant value is expected except for the adsorption sites near the edge of the platelet.
Full coverage, i.e. attaching H2 on both sides of the PAH, shows the same trend.
The molecule with the highest occupation and largest binding energy per H2 is coronene with alternating occupation of the
outer rings on both sides. (see Fig. 2.6)
neighbour ring.
These observations are in agreement with Diep's and Johnson's study of the H2-H2 interaction [54,55].
Therefore, all geometrical models with several H2 molecules at a PAH side have the neighbouring rings of an H2 physisorption site uncovered.
Considering the above mentioned relations, one H2 molecule can be attached to each side of benzene and naphthalene (one at each side), two to each anthracene side, and three to each side of pentacene, triphenylene and coronene.
Table 2.2 shows that the total physisorption energy per H2 is the average of the individual physisorption energies of the occupied sites.
In other words, attaching H2 molecules at a PAH side is a completely additive process, and for a graphene sheet a constant value is expected except for the adsorption sites near the edge of the platelet.
Full coverage, i.e. attaching H2 on both sides of the PAH, shows the same trend.
The molecule with the highest occupation and largest binding energy per H2 is coronene with alternating occupation of the
outer rings on both sides. (see Fig. 2.6)
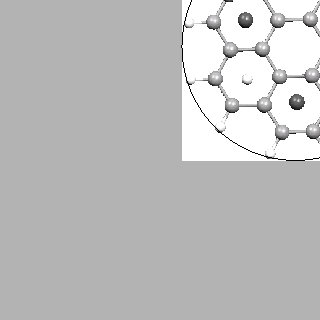
Figure 2.6:
Coronene model structure with physisorbed hydrogen at the energetically favoured pattern.
On the right side, an estimated nanostructured graphene layer with the same physisorption pattern is
displayed. The coronene model side is indicated by a ring.
 |
This gives a physisorption energy of 6.0 kJmol per H for each absorption site at the outer rings of coronene, the same value as the physisorption energy of a single H at the same site.
Thus for graphene, it is expected that the interaction should increase to a value a bit higher then 7.2 kJmol per H, as estimated using Eq. 2.1 and the model with a single hydrogen molecule.
In agreement with DFT calculations [52], the above presented computations show that the normal orientation of H to the PAH gives the strongest interaction, whereas in the parallel orientation, the stacking is by 1.2 kJmol weaker.
However, these physisorption energies hold only for H2 PAHs models where the physisorption depends on the orientation of H2 molecule and the temperature dependence is neglected.
Lyuben Zhechkov
2007-09-04