H
 C
C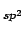 Potential
Potential
The contributions from the hexagon ring of infinite graphene platelet to the total potential energy surface are equivalent.
Therefore it can be considered that the contribution of each sp carbon atom is six times smaller than the contribution of a six member ring.
Nevertheless, direct ab initio evaluation of the sufficiently detailed potential energy surface of a graphene platelet is prohibitively expensive.
Instead, the H2graphene PES is described not by the equation 2.1, but by the exp
carbon atom is six times smaller than the contribution of a six member ring.
Nevertheless, direct ab initio evaluation of the sufficiently detailed potential energy surface of a graphene platelet is prohibitively expensive.
Instead, the H2graphene PES is described not by the equation 2.1, but by the exp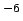 form of the Lenard-Jones (LJ) H2C pair (see Eq. 1.14), where
form of the Lenard-Jones (LJ) H2C pair (see Eq. 1.14), where 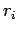 is the distance between H2 centre of mass and carbon atom
is the distance between H2 centre of mass and carbon atom 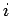 .
Hence, the evaluation of the interaction potential for graphene platelet turns simply to a sum over all the carbon centres:
.
Hence, the evaluation of the interaction potential for graphene platelet turns simply to a sum over all the carbon centres:
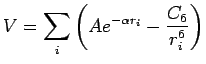 |
|
|
(2.2) |
The parameters  ,
,  , and
, and
 are fitted to reproduce the MP2/cc-pVTZ results of the
are fitted to reproduce the MP2/cc-pVTZ results of the 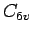 symmetric
H2benzene and H2coronene model system (see 2.4).
symmetric
H2benzene and H2coronene model system (see 2.4).
1.0
Table 2.4:
Van der Waals potential parameters, fitted to benzene MP2/cc-pVTZ and CCSDT(T)/cc-pVTZ, and coronene MP2/cc-pVTZ.
| PAH/method |
A, kJ/mol |
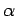 |
, kJ/mol |
| benzene/MP2 |
58783 |
-4.7015 |
2583 |
| benzene/CCSD(T) |
52657 |
-4.6827 |
2018 |
| coronene MP2 |
106143 |
-5.0082 |
1676 |
Such a description is more accurate and more flexible than the one described in section 2.2.
It gives the possibility to evaluate more accurate potentials not only for large planar structures, but also for bent carbon surfaces as in carbon nanotubes and fullerenes.
As can be seen from Fig. 2.7 the parameters give very good reproduction of the ab initio results.
Moreover, a crosscomparison of the potentials shows that the parameters fitted for H2-coronene complex gives a `H2C6H6' curve which lies very close to the Coupled Cluster `H2C6H6' values (see Fig. 2.7).
In other words, besides the reasonable performance of the the MP2/cc-pVTZ level of theory, when enlarging the model system (e.g adding more atoms to the model) a better description of the potential interaction can be achieved.
Figure:
Results and details of quantum-chemical calculations of
H2 adsorbed at the C site of benzene. Interaction energies for
selected ab initio methods with and without counterpoise BSSE corrections,
and performance of the parametrised Lenard-Jones potentials.
site of benzene. Interaction energies for
selected ab initio methods with and without counterpoise BSSE corrections,
and performance of the parametrised Lenard-Jones potentials.
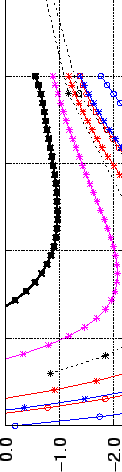 |
On the other hand the fitted potential slightly underestimates (by 0.4 kJ mol
mol ) the translational
barriers between two adjacent minima (Fig. 2.8).
) the translational
barriers between two adjacent minima (Fig. 2.8).
Figure:
Ab initio computations (stars) compared to the parametrised LJ
potential (solid lines) of H2 interacting with coronene. (Left) Movement of H2
perpendicular to coronene along the C axis. (Right) Movement of H2 toward
an atom (blue) and toward a bond (red) at a distance between molecular plane and centre
of H2 of 3.1 Å. Note the difference in the energy scales in Left and Right
|
However, this deviation is small compared with 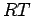 and is therefore not expected to affect the thermodynamical properties.
The three types of parameters (one fitted to benzene CCSD(T)/cc-pVTZ and the others fitted to coronene and benzene MP2/cc-pVTZ calculations) have been compared with an empirical potential.
and is therefore not expected to affect the thermodynamical properties.
The three types of parameters (one fitted to benzene CCSD(T)/cc-pVTZ and the others fitted to coronene and benzene MP2/cc-pVTZ calculations) have been compared with an empirical potential.
Figure:
Comparison of empirical and ab initio parameters for
H2C interaction potential.
|
In Figure 2.9, it is shown that they slightly overestimate the experimental values.
Considering the fact that the ab initio calculations completely neglect the thermodynamical conditions of the experiment in general, the fits may be taken as realistic.
Therefore the so derived parameters are used further (see Chapter 1) to evaluate periodic graphene-like structures with experimental or quantum mechanically computed CC bond lengths (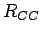 ).
).
Lyuben Zhechkov
2007-09-04