Isolated SWCNT
The major aim of the present simulations is to demonstrate how the curvature of the concave part of the tube influences the physisorption of molecular hydrogen.
In this approach the contribution from the outer convex surface of the nanotube is completely neglected.
Since the box used in the periodic boundary conditions is rectangular and the tube has cylindrical symmetry, additional restrictions to the model have been made.
In order to remove any probability of binding states outside the tube, in this area the interaction potential has been set up to be always repulsive.
The strongest interaction inside a nanotube is found to have a value (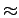 25 kJ
25 kJ mol
mol ) 40 % higher than the one in the graphene and CIG models ( 15 kJmol) (see respectively Fig. 3.1, 4.1 and 5.1).
) 40 % higher than the one in the graphene and CIG models ( 15 kJmol) (see respectively Fig. 3.1, 4.1 and 5.1).
Figure:
Scan of the interaction potential along the diameter to different
SWCNTs including only armchair and zigzag configurations.
The legend gives the radius of the narrowest, the widest and the nanotube with the strongest
interaction potential. The Hamada indices are given in brackets. Zero on the abscissa coincides with the tube axis
|
The highest values of the interaction potential are calculated for nanotubes with diameters between 6.2 and 8.6 Å, whereas below 5.5 Å the potential becomes strongly repulsive.
A comparison with hypothetical double layer structures (see Chapter 3.1) demonstrates that maxima for both structures (nanotubes and slit graphene pores) are found for nearly the same pore width.
While the slit pore is a space confined between two graphene sheets, the nanotube can be considered as quasi-one dimensional cavity confined by the enrolment of a single layer.
Therefore, compared with the double layer graphene model, the maximal H2 nanotube interaction along its axis can be attributed to the equidistant contributions from the carbon atoms.
Since the H2carbon pair interaction is additive (see Chapter 2), favourable geometrical properties can be easily tuned in order to control the interaction potential.
One of the consequences is that a nanotube with extremely big diameter will approach of interaction potential produced by a plane surface.
For example, the interaction along the tube axis increases rapidly for diameters
nanotube interaction along its axis can be attributed to the equidistant contributions from the carbon atoms.
Since the H2carbon pair interaction is additive (see Chapter 2), favourable geometrical properties can be easily tuned in order to control the interaction potential.
One of the consequences is that a nanotube with extremely big diameter will approach of interaction potential produced by a plane surface.
For example, the interaction along the tube axis increases rapidly for diameters 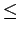 8.6 Å and then starts slowly to approach zero.
On the other hand, the potential in the vicinity of the tube wall approaches to a value characteristic for the surface of graphene layer
(7 kJmol, see and section 2.2).
This effect is demonstrated in Figure (5.1)0.55.1, which can be considered as diametral slice of the 3D-potential along the tube axis.
8.6 Å and then starts slowly to approach zero.
On the other hand, the potential in the vicinity of the tube wall approaches to a value characteristic for the surface of graphene layer
(7 kJmol, see and section 2.2).
This effect is demonstrated in Figure (5.1)0.55.1, which can be considered as diametral slice of the 3D-potential along the tube axis.
Regardless of the relatively high values computed for the inner tube interaction potential, the entropy still has significant contribution and strongly affects the interaction free energy 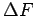 .
A comparison between the /
.
A comparison between the /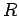 correlations0.55.2 for nanotube and graphenegraphene models shows no significant difference (see Fig. 3.5 and 5.2 Right).
correlations0.55.2 for nanotube and graphenegraphene models shows no significant difference (see Fig. 3.5 and 5.2 Right).
Figure:
Equilibrium constants 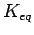 (Left) and reaction free energies
(Right) are plotted in kJmol as a function of the tube radius at T = 300 K.
(Left) and reaction free energies
(Right) are plotted in kJmol as a function of the tube radius at T = 300 K.
|
The concavely bent nanotube surface increases the gas-host binding by only 2 kJmol at 300 K.
In other words, the interaction free energies for different materials have the same values as far as the pore width is comparable.
However, the corresponding equilibrium constants, which represent the equilibrium ratio between the adsorbed and disrobed (H2_ads/H2_des) molecules, show that carbon nanotubes have an order of magnitude larger equilibrium constants (see Fig. 3.5 and 5.2 Left).
Such a behaviour of can be better understood in terms of density of states (DOS).
The comparison between layered a graphene system (Fig. 3.6, interlayer distance d=8 Å) and a carbon nanotube having similar pore width (Fig. 5.3, diameter d8 Å)
Figure:
Density of states (DOS) of molecular hydrogen in a nanotube
with Hamada indices 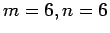 with diameter 8 Å (
with diameter 8 Å (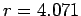 Å) and a potential-free simulation box.
Å) and a potential-free simulation box.
|
shows dramatic a decrease in the binding states density.
On the other hand, the energetically lowest binding states in the nanotube have binding energies which are twice ``deeper'' than those of the graphene system.
Thus, despite the comparable interaction free energy () for those systems, the core of a nanotube with its positively bent wall may considerably increase the hydrogen ``trapping'' capability at 300 K.
Although, the temperature dependence of the interaction free energy and the corresponding equilibrium constants shows similar trends (like the double layered graphene system), the slope of the and curves is more pronounced.
For example, the interaction free energy change in the range of 150 to 300 K is more than 3 kJmol for the nanotubes, (see Fig. 5.4) whereas the change for a slit pore with the same width is 0.5 kJmol(see Fig. 3.4).
This indicates that small nanotube channels, despite their lower energy binding states, loose very fast their sorption abilities with the increase of the temperature.
As can be seen on Figure 5.4 the smaller the radius of the tube, the faster the values diminish.
In other words, it is easier to ``freeze'' H2 inside a narrow nanotube, but small increase in the temperature make it easier to desorb.
Figure:
Equilibrium constants (Left) and reaction free energies
(Right) are plotted in kJmol as a function of the temperature for nanotubes with diameter 8 Å.
|
The data, presented so far, suggests that only very few nanotubes with certain diameters can effectively increase hydrogen gas density in their cavities.
Too wide nanotubes are expected to behave similarly as the single graphene sheet model.
The narrow nanotubes tubes (d7 Å) on the other hand can not accommodate effectively H2 molecules[86].
Therefore, a reasonable pore width in H2 storage material is considered to be 7d10 Å (where ``d'' is the nanotube diameter).
Such a suggestion can be easily explained by simple geometrical considerations.
If the H2host interaction is considered as a sum of H2-C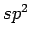 pair interactions, then the total interaction will depend on the attractive surface (density of carbon atoms per unit of area) and the H2-surface distance.
Since the inner surface of the narrow nanotubes have a positive curvature, the overlapping of the pair potentials is much denser.
In the wider cylinders the strongest overlapping covers only the inner surface of the nanotube.
Therefore, the strongest interaction in the nanotubes with diameter
pair interactions, then the total interaction will depend on the attractive surface (density of carbon atoms per unit of area) and the H2-surface distance.
Since the inner surface of the narrow nanotubes have a positive curvature, the overlapping of the pair potentials is much denser.
In the wider cylinders the strongest overlapping covers only the inner surface of the nanotube.
Therefore, the strongest interaction in the nanotubes with diameter 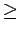 9 Å is located on the tube walls and slowly approaches the values for graphene sheet with the straightening of the of the tube wall.
For the very wide tubes the core interaction potential (around the axis) has values even smaller than the suggested entropy contribution to the interaction energy (see Chapter 2.5).
The effect can be easily noticed on Fig. 5.1, where the interaction potential in the vicinity of the nanotube (with diameter 9 Å) wall is similar to the one for graphite surface (7 kJmol).
A more accurate interpretation can be done in terms of physisorbed gas densities.
Figure 5.5 presents the volumetric capacities together with the corresponding gravimetric capacities
for different external pressures and temperatures.
9 Å is located on the tube walls and slowly approaches the values for graphene sheet with the straightening of the of the tube wall.
For the very wide tubes the core interaction potential (around the axis) has values even smaller than the suggested entropy contribution to the interaction energy (see Chapter 2.5).
The effect can be easily noticed on Fig. 5.1, where the interaction potential in the vicinity of the nanotube (with diameter 9 Å) wall is similar to the one for graphite surface (7 kJmol).
A more accurate interpretation can be done in terms of physisorbed gas densities.
Figure 5.5 presents the volumetric capacities together with the corresponding gravimetric capacities
for different external pressures and temperatures.
Figure:
Volumetric (Top) and gravimetric (Bottom) storage capacities for
interior of the nanotubes with radius are given for various temperatures (see the legend). Left and right columns give the values
respectively for pressures at 5 and 10 MPa.
The symbols on the curves denote that the approximation is within the limits (pressure and temperature) of the real gas
equation of state [36]. The targets[4] automotive
applications (Gwt) 6.0 %,(V) 44.4 cm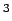 /mol) are indicated as horizontal lines.
/mol) are indicated as horizontal lines.
|
As can be noticed, the increase of both temperature and pressure improves the storage abilities.
However, while the gravimetric capacities decrease smoothly with broadening the pore radius the density of stored hydrogen gas is expected to drop down exponentially.
This trend is even more pronounced for higher temperatures and demonstrates that the abundance of hydrogen gas at ambient temperatures is strongly controlled by the pore size.
The lowering of the H2-carbon mass ratio may be compensated to some extend by the decrease of the total carbon density.
Hence, the decrease in the gravimetric storage capacity at 300 K becomes negligible (see Fig. 5.5).
Nonetheless, these numbers do not consider the outer surface or the interstitial space between nanotubes.
Therefore, these values should be considered as giving information only about the trends for the H2 storage abilities of positively curved surfaces.
The more complex models as bundles of nanotubes and carbon foams are discussed in the next sections and chapters.
Lyuben Zhechkov
2007-09-04